Thalassemia
Mediterranean anemia, Cooley’s anemia, and Cooley anemia are other names for thalassemia. Because it was first discovered in communities residing close to the Mediterranean Sea, the term is derived from the Greek word “thalassa” (meaning sea). Over generations, Cooley’s anemia has affected millions globally, a genetic disorder that affects blood and hemoglobin production. You might wonder about its origins and how it manifests. This condition, characterized by reduced or absent hemoglobin, leads to anemia and other complications, necessitating a clear understanding of its causes, symptoms, and potential treatment approaches.
Key Takeaways:
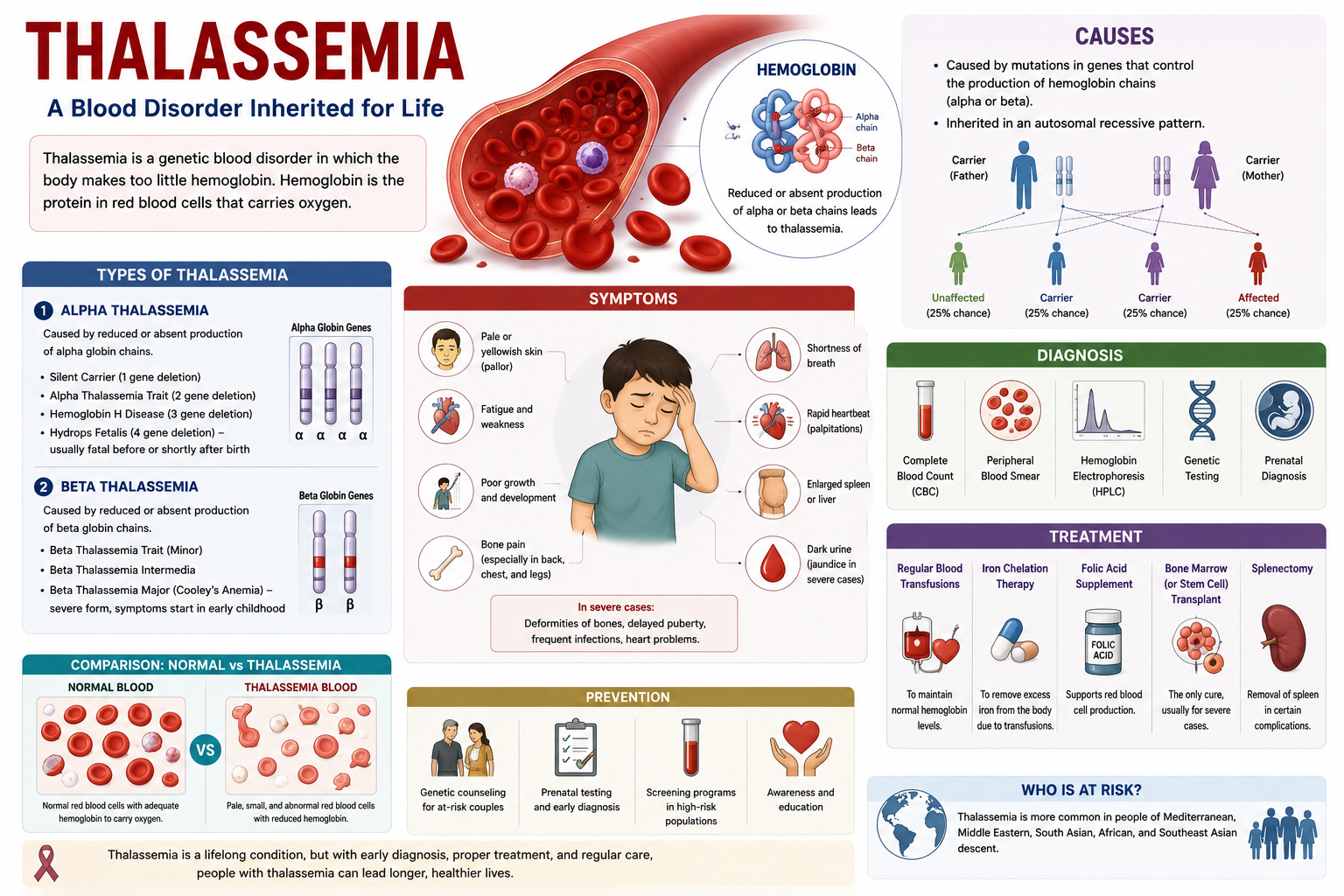
- Thalassemia is a genetic blood disorder characterized by the body producing an abnormal form or inadequate amount of hemoglobin, the protein in red blood cells that carries oxygen. This deficiency leads to anemia, which can range from mild to severe depending on the specific type and inheritance pattern.
- The condition is inherited in an autosomal recessive pattern, meaning an individual must inherit two copies of a mutated gene (one from each parent) to develop the more severe forms of Mediterranean anemia. Carriers, who inherit only one mutated gene, often experience milder symptoms or none at all, a condition known as thalassemia trait.
- There are two main types, alpha-thalassemia and beta-thalassemia, categorized by the specific globin protein chain affected in the hemoglobin molecule. Each type has varying degrees of severity, from asymptomatic carriers to life-threatening conditions like Cooley’s anemia (severe beta-thalassemia).
- Common symptoms include fatigue, weakness, pale skin, stunted growth, dark urine, and jaundice, all stemming from the body’s inability to produce healthy red blood cells and transport oxygen efficiently. Bone deformities, particularly in the face, can also occur in severe, untreated cases due to bone marrow expansion.
- Conventional medical management frequently involves regular blood transfusions to provide healthy red blood cells, especially for individuals with thalassemia major. Iron chelation therapy is often necessary to remove excess iron that accumulates from these transfusions and prevent organ damage.
- Homeopathic treatments aim to support the body’s vital force and improve overall health, potentially mitigating some symptoms and reducing the frequency of conventional interventions in milder cases. These remedies are highly individualized, selected based on a patient’s unique symptom presentation and constitutional type.
- While homeopathic approaches can offer supportive care, they are generally considered complementary and not a standalone cure for severe forms of thalassemia, which often require lifelong medical supervision and intervention. Consultation with both conventional and homeopathic practitioners is advisable to develop a comprehensive care plan.
Cooley’s anemia: Genetic Etiology and Patterns of Inheritance
Understanding Genetic Mutations
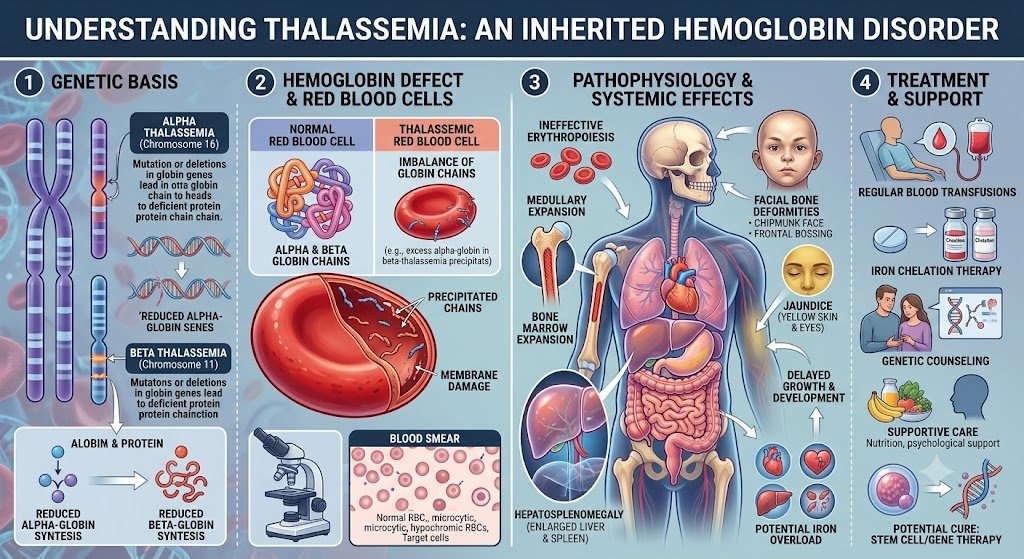
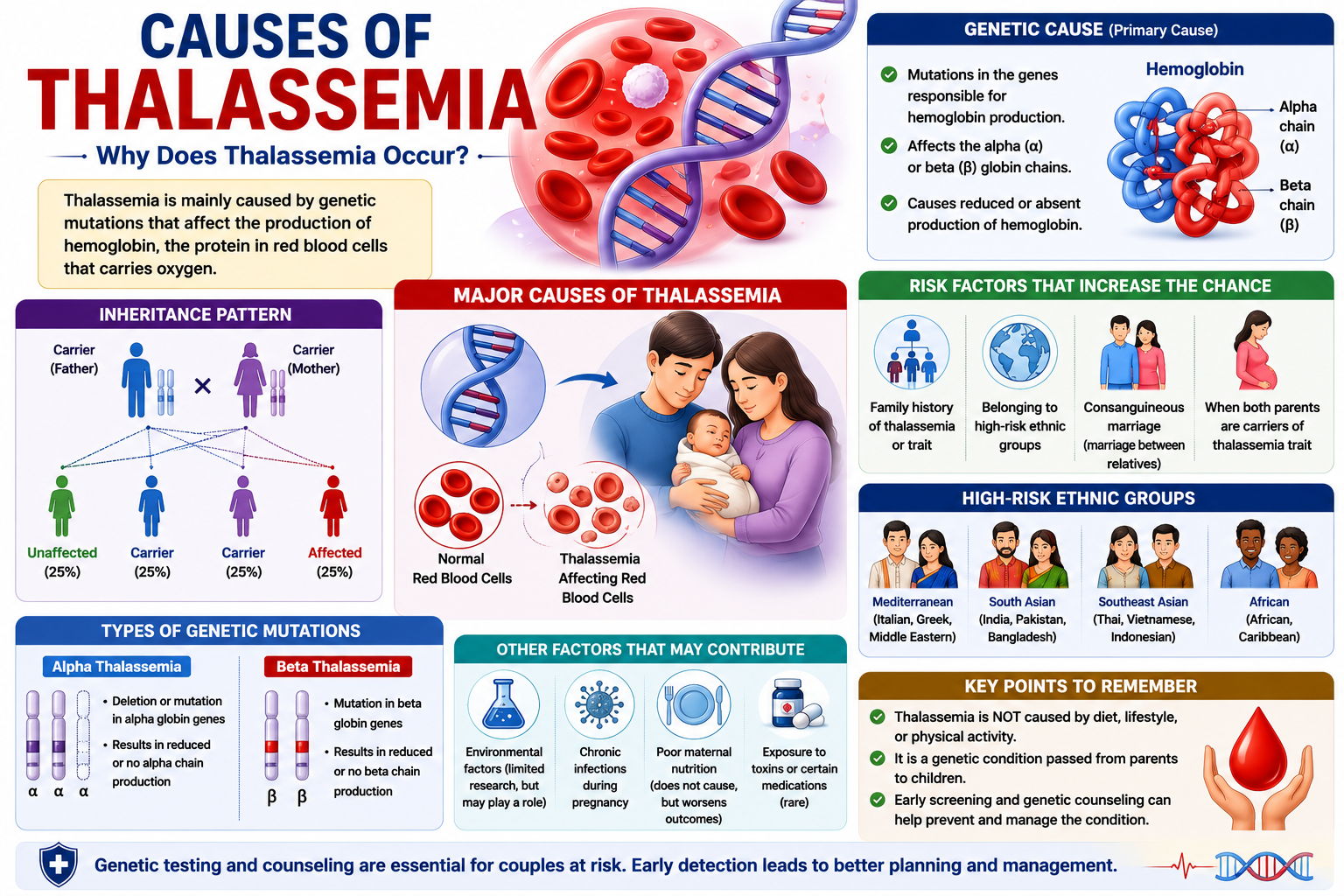
Thalassemia originates from specific genetic mutations that disrupt the normal production of hemoglobin, the protein in red blood cells responsible for oxygen transport. These mutations primarily affect either the alpha-globin or beta-globin genes, leading to reduced or absent synthesis of one of the globin chains. The specific gene affected determines whether an individual develops alpha-thalassemia or beta-thalassemia, each with varying degrees of severity.
Each form of thalassemia stems from distinct genetic alterations. For instance, alpha-thalassemia typically involves deletions of one or more of the four alpha-globin genes located on chromosome 16. Beta-thalassemia, in contrast, usually results from point mutations in the two beta-globin genes situated on chromosome 11, which can lead to various effects, from reduced production to complete absence of the beta chain.
Identifying the precise genetic mutation is often crucial for accurate diagnosis and prognosis. Genetic testing can pinpoint the exact alteration, allowing healthcare providers to differentiate between the numerous subtypes and predict the potential clinical course. This detailed genetic understanding helps in counseling affected families about the inheritance risks.
Hereditary Patterns and Family History
Thalassemia follows an autosomal recessive pattern of inheritance. This means that for a child to be affected with a severe form of Mediterranean anemia, they must inherit two copies of the mutated gene, one from each parent. If an individual inherits only one copy of the mutated gene, they are considered a carrier (also known as having thalassemia trait) and typically do not exhibit significant symptoms, though they can pass the gene on to their offspring.
Parents who are both carriers of the Cooley’s anemia gene face a 25% chance with each pregnancy that their child will inherit two copies of the mutated gene and thus develop a severe form of the disorder. There is also a 50% chance that their child will be a carrier like them, and a 25% chance that their child will inherit two normal genes and be unaffected. Understanding these probabilities is fundamental for genetic counseling.
Family history plays a significant role in identifying individuals at risk. If thalassemia or a carrier state is known to exist within a family, particularly in populations where the condition is more prevalent (such as individuals of Mediterranean, South Asian, African, or Southeast Asian descent), genetic screening becomes especially important. Early identification through family history allows for informed reproductive decisions and timely medical intervention if necessary.
Considering the autosomal recessive nature, a thorough family medical history can reveal patterns of inheritance that might otherwise go unnoticed. For example, if several relatives on both sides of a family have a history of anemia or have been identified as carriers, it strengthens the likelihood that a couple may both carry the gene, increasing their risk of having an affected child. This information can prompt further genetic testing for prospective parents.
Classification of Thalassemia Types
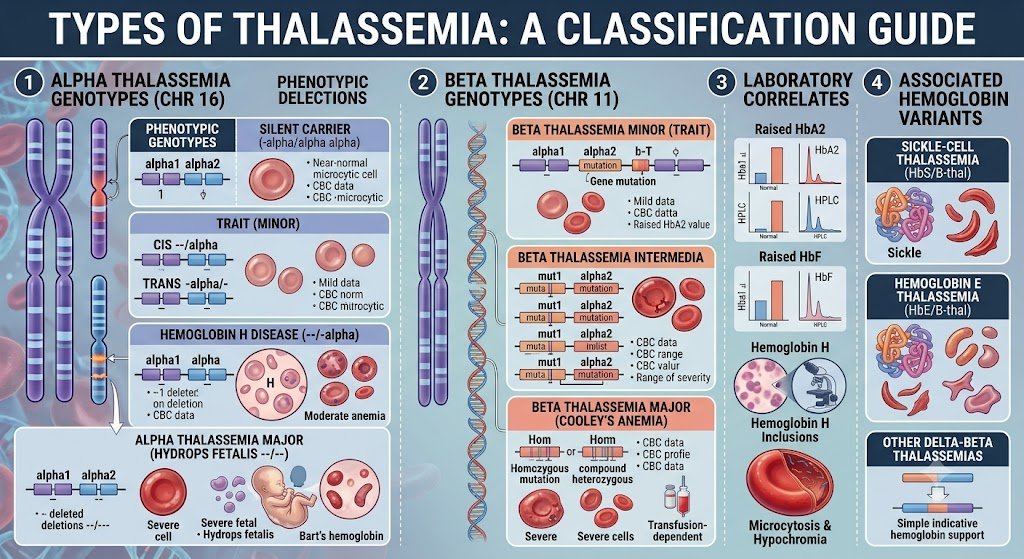
Mediterranean anemia presents in various forms, primarily based on which globin protein chain is affected. Understanding these distinctions is imperative for accurate diagnosis and management. The two main classifications are alpha thalassemia and beta thalassemia, named after the specific globin chains whose production is reduced or absent. One can further break down these primary categories based on the severity of the genetic mutation and its phenotypic expression. For instance, within both alpha and beta thalassemia, you will encounter terms such as thalassemia minor, thalassemia major, and thalassemia trait. These terms describe a spectrum from asymptomatic carriers to individuals requiring lifelong medical intervention. This classification system helps clinicians predict the likely course of the disease and tailor treatment strategies accordingly.
| Alpha Thalassemia | Reduced or absent production of alpha globin chains. |
| Beta Thalassemia | Reduced or absent production of beta globin chains. |
| Thalassemia Minor | The mildest form, often asymptomatic, is also called the thalassemia trait. |
| Thalassemia Major | This variant is the most severe form, requiring regular blood transfusions and extensive medical care. |
| Thalassemia Trait | Individuals carry one defective gene, usually exhibiting mild anemia or no symptoms. |
Distinguishing alpha and beta variants
The fundamental difference between alpha and beta thalassemia lies in the specific globin chain that is insufficiently produced. Alpha thalassemia results from mutations affecting the alpha-globin genes, located on chromosome 16. Humans typically possess four alpha-globin genes, two inherited from each parent. Beta thalassemia, on the other hand, arises from mutations in the beta-globin gene, found on chromosome 11. Individuals normally have two beta-globin genes, one from each parent. The impact of these genetic variations directly correlates with the number of affected genes. This distinction in the genetic locus and the specific protein chain involved dictates the molecular pathology and often the clinical presentation.
Severity levels from trait to major disease
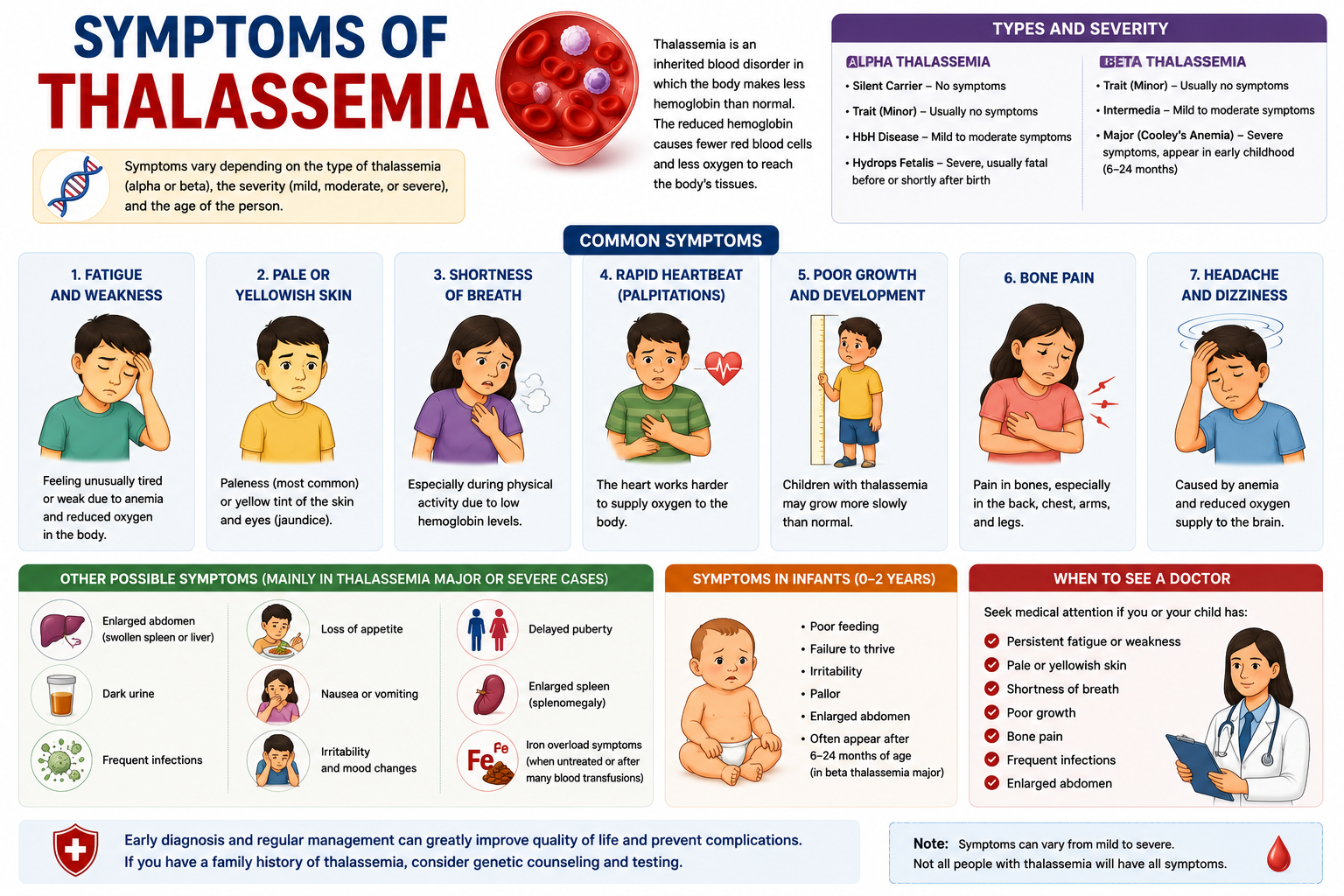
The severity of thalassemia spans a broad range, from an asymptomatic carrier state to a life-threatening condition. Thalassemia trait, also known as thalassemia minor, represents the mildest form where an individual carries only one mutated gene, either alpha or beta. These individuals typically experience no symptoms or very mild anemia, often detected incidentally. Moving along the spectrum, more significant genetic defects lead to more pronounced clinical manifestations. Thalassemia major, for example, is the most severe form, resulting from inheriting two mutated genes, one from each parent. Individuals with thalassemia major face severe anemia, requiring lifelong blood transfusions and intensive medical management to survive. This spectrum highlights how the number of affected genes directly correlates with the disease’s impact on an individual’s health. The intermediate forms, sometimes referred to as thalassemia intermedia, present with symptoms that are more severe than thalassemia minor but less severe than thalassemia major. These patients might require occasional blood transfusions, particularly during times of stress or infection, but not as frequently as those with thalassemia major. The clinical picture varies widely, depending on the specific genetic mutations involved. This variability makes careful genetic counseling and monitoring important for affected families.
Standard Medical Management of Thalassemia
Clinical protocols for blood management
Regular blood transfusions form the cornerstone of standard medical management for individuals with severe forms of thalassemia, such as beta-thalassemia major. These transfusions aim to maintain adequate hemoglobin levels, typically above 9-10 g/dL, and help suppress ineffective erythropoiesis, as well as prevent many complications associated with chronic anemia, including growth retardation and bone deformities. The frequency of these transfusions varies based on the individual’s specific needs and the severity of their condition. Maintaining a consistent transfusion schedule is critical for preventing the symptoms of severe anemia, which can significantly impact quality of life. Patients often receive transfusions every two to four weeks, depending on their rate of red blood cell destruction and bone marrow suppression. This regimen ensures a continuous supply of healthy red blood cells, reducing fatigue, improving energy levels, and supporting normal development in pediatric patients. You will find that careful monitoring of blood parameters, including hemoglobin levels and iron overload indicators, guides the transfusion strategy. Pre-transfusion and post-transfusion blood tests help clinicians tailor the volume and frequency of transfusions to each patient, optimizing therapeutic benefits while minimizing potential risks. This personalized approach is imperative for long-term management.
Addressing iron toxicity and chelation
Blood transfusions, while life-saving, introduce a significant challenge: iron overload. Each unit of transfused red blood cells contains approximately 200-250 mg of iron, and the body has no natural mechanism to excrete excess iron. This accumulation leads to hemosiderosis, where iron deposits in vital organs such as the heart, liver, and endocrine glands, causing progressive damage and organ dysfunction. Iron chelation therapy is therefore indispensable for patients undergoing chronic blood transfusions. These medications work by binding to excess iron in the bloodstream, forming a compound that can be excreted from the body, primarily through urine and feces. Initiating chelation therapy early, often after 10-20 transfusions or when serum ferritin levels exceed 1000 ng/mL, is crucial to prevent irreversible organ damage. Several chelation agents are available, each with different administration routes and side effect profiles. Deferoxamine, administered subcutaneously or intravenously over 8-12 hours, was historically the primary chelator. Oral options, such as deferiprone and deferasirox, have greatly improved patient convenience and compliance, allowing for more flexible treatment regimens. Your doctor will determine the most appropriate chelator or combination of chelators based on your individual iron burden, organ function, and tolerance. Patients on chelation therapy require regular monitoring of their iron levels, including serum ferritin, liver iron concentration (LIC) measured by MRI, and cardiac T2* MRI, to assess the effectiveness of the treatment and adjust dosages as needed. This ongoing assessment helps prevent both under-chelation, which can cause more organ damage, and over-chelation, which can lead to its own complications.
Homeopathic Treatment for Thalassemia
The goal of homeopathic treatment for thalassemia is to provide patients with this genetic blood illness with tailored, supportive care that strives to control recurrent symptoms, improve general well-being, and increase quality of life. Thalassemia impacts hemoglobin production, often leading to weakness, exhaustion, and the need for constant medical supervision. In addition to traditional treatment, homeopathy uses a customized strategy to support the body’s natural equilibrium, assisting patients in better managing their symptoms and maintaining their general health.
To finally lessen the necessity for repeated blood transfusions, homeopathy treats the underlying problem and provides medication. Additionally, homeopathic remedies help strengthen the immune system, which reduces the frequency of respiratory infection episodes. When it comes to thalassemia, homeopathic medicine plays an additional role.
According to some recent research, homeopathic treatments may help manage the symptoms of thalassemia. They assert that these treatments may strengthen the immune system, improve general health, and strengthen the body’s capacity to manage symptoms such as anemia. Additionally, other clinical studies showed that patients who utilized homeopathy needed fewer blood transfusions than those who did not.
Some patients may decide to utilize homeopathic remedies as a supplemental strategy to help manage mild symptoms or enhance general health, even if they do not replace traditional medical therapies for thalassemia. Before contemplating it, patients should always speak with their homeopathic physician to ensure that their illness is managed safely and effectively.
Conclusion
You now understand that thalassemia, a genetic blood disorder, stems from inherited defects in hemoglobin synthesis. The severity of your condition, whether you have alpha or beta thalassemia, major or minor, dictates the specific symptoms you might experience, ranging from mild anemia to severe, life-threatening complications requiring regular transfusions. Your genetic makeup, inherited from your parents, ultimately determines the type and extent of the disease you present with, underscoring the importance of genetic counseling for affected families. Managing your Mediterranean anemia effectively often involves a multi-pronged approach tailored to your specific needs. While conventional medical treatments like blood transfusions and chelation therapy remain cornerstones for severe forms, you may also explore complementary strategies. Homeopathic treatments, for example, aim to stimulate your body’s self-regulatory mechanisms, potentially offering supportive care alongside your primary medical regimen. Always discuss any alternative therapies with your healthcare provider to ensure they complement, rather than conflict with, your established medical plan. Living with thalassemia requires ongoing attention to your health and a proactive approach to your care. Regular medical follow-ups, adherence to prescribed treatments, and a healthy lifestyle are imperative components of managing the condition effectively. You can maintain a good quality of life by staying informed about your specific type of thalassemia and actively participating in decisions regarding your treatment path, working closely with your medical team to address any challenges that arise.
FAQ
Q: What is thalassemia?
A: Thalassemia represents a group of inherited blood disorders characterized by the body producing an insufficient amount of hemoglobin, the protein in red blood cells responsible for carrying oxygen. This deficiency leads to anemia, which can range from mild to severe, depending on the specific type and genetic mutation involved. Normal red blood cells have a lifespan of about 120 days, but in individuals with thalassemia, these cells are often fragile and have a much shorter life, sometimes lasting only a few weeks.
Q: What causes thalassemia?
A: Genetic mutations are the root cause of thalassemia, affecting the genes responsible for producing hemoglobin. Hemoglobin consists of two main types of protein chains: alpha and beta. Mutations in the genes controlling the production of alpha globin chains lead to alpha-thalassemia, while mutations in the genes controlling beta globin chains result in beta-thalassemia. These genetic alterations disrupt the delicate balance required for proper hemoglobin synthesis, impairing the red blood cells’ oxygen-carrying capacity.
Q: How is thalassemia inherited?
A: Thalassemia follows an autosomal recessive inheritance pattern. This means that an individual must inherit two copies of the mutated gene, one from each parent, to develop the more severe forms of the disorder, such as thalassemia major. If a person inherits only one copy of the mutated gene, they are considered a carrier (also known as having thalassemia minor or thalassemia trait). Carriers typically experience no symptoms or very mild anemia, but they can pass the gene to their children. For example, if two carriers have a child, there is a 25% chance with each pregnancy that the child will inherit two mutated genes and develop a severe form of thalassemia.
Q: What are the main types of thalassemia?
A: The primary classifications of thalassemia are alpha-thalassemia and beta-thalassemia, named after the specific globin protein chain affected. Alpha-thalassemia occurs when there are defects in the genes coding for the alpha globin protein; its severity depends on how many of the four alpha globin genes are mutated. Beta-thalassemia results from mutations in the genes coding for the beta globin protein; it is further categorized into beta-thalassemia major (severe), beta-thalassemia intermedia (moderate), and beta-thalassemia minor (carrier state). Each type presents with a distinct clinical picture and varying degrees of anemia.
Q: What are the common symptoms of Mediterranean anemia?
A: Symptoms of thalassemia vary significantly based on the type and severity. Individuals with thalassemia minor often experience no symptoms or mild fatigue. Those with more severe forms, such as thalassemia major, typically present with symptoms within the first two years of life. Common signs include severe fatigue, weakness, pale skin (pallor), slow growth, abdominal swelling due to an enlarged spleen or liver, and dark urine. Bone deformities, particularly in the face, can also develop over time due to the bone marrow expanding to produce more red blood cells.
Q: What are the standard medical treatments for thalassemia?
A: Standard medical management for severe thalassemia primarily involves regular blood transfusions to provide healthy red blood cells and alleviate anemia. Patients receiving frequent transfusions often develop an excess of iron in their bodies, a condition called iron overload, which can damage organs. Iron chelation therapy, using medications to remove excess iron, becomes a critical component of treatment to prevent complications. In some cases, a bone marrow transplant (also known as a stem cell transplant) can offer a potential cure, particularly for younger patients with a suitable donor.
Q: Can homeopathy offer treatment for thalassemia?
A: Homeopathic treatment for thalassemia aims to support the individual’s overall health and manage symptoms, particularly in milder cases like thalassemia minor. Remedies are selected based on the patient’s unique constitutional symptoms rather than solely on the diagnosis of thalassemia itself. For example, a homeopath might consider remedies such as Ferrum metallicum for anemia and fatigue or Phosphorus for issues related to bleeding tendencies, always tailoring the prescription to the individual’s specific presentation. Homeopathy is generally considered a complementary approach and should not replace conventional medical treatments, especially for severe forms of thalassemia that require regular transfusions and chelation therapy.
Homeopathic Treatment for Thalassemia in Philadelphia
At the Philadelphia Homeopathic Clinic, Victor Tsan, MD, and associates cure different forms of anemia, including Mediterranean anemia, using various holistic techniques, including homeopathy, acupuncture, and medicinal herbs. Contact our office or use our online scheduling system to make an appointment for the holistic evaluation and discuss with Dr. Tsan whether alternative medicine is a good treatment option for you.